To this point in our study of chemistry, we have been concerned only with the composition of the equilibrium mixture, not the length of time required to obtain it. However, the time required is also an important consideration. If a reaction proceeds too slowly, it will not be useful no matter how large the equilibrium constant, and if a reaction proceeds too rapidly, it may not be possible to control it. Thermodynamics allows us to predict the extent of a reaction, but it tells us nothing about the speed of the reaction or how it occurs. These latter two considerations are the domain of kinetics, the study of the rates and mechanisms of chemical reactions.
10.1. Reaction Rates
Introduction
Rates are changes in one quantity with respect to another, and reaction rates are measured as changes in concentration with respect to time. They are the most important of the kinetic parameters, and we begin our study of kinetics with a treatment of reaction rates.
Objectives
•
Express the relative rates of consumption of reactants and formation of products from the coefficients in a balanced chemical reaction.
10.1-1. Concentrations versus Time
It takes time for the concentrations of reactants and products to change from their initial to their equilibrium values, but once equilibrium is established, the concentrations no longer change. Kinetics studies the reaction during the time of change, while thermodynamics is concerned only with the initial and equilibrium conditions. Thus, the region of time in which the concentrations are changing is called the kinetic region, while the region after equilibrium is established is called the thermodynamic region.Consider the decomposition of 0.020 M HI at 700 °C, 2 HI(g) → H2(g) + I2(g). The concentrations of both reactants and products are shown in Figure 10.1a as a function of time in seconds. The concentration of HI decreases for about 10,000 seconds (about three hours) and then levels off at 0.015 M. During that time, the concentrations of H2 and I2 each rise from zero to about 0.0025 M. Once the concentrations are no longer changing, the system has reached equilibrium. Thus, approximately three hours are required to obtain an equilibrium mixture at this temperature. Δng = 0 for the reaction, so the thermodynamic equilibrium constant for the reaction is equal to Kc, so it can be determined from the equilibrium concentrations,
K =
[H2][I2]
[HI]2
=
(0.0025)(0.0025)
(0.015)2
= 0.028
Figure 10.1a: Concentrations versus Time for the Decomposition of HI at 700 °C
If the reaction were extensive, 0.020 M HI would react to produce a mixture that was 0.010 M in each H2 and I2, and we now examine the reaction from the reverse direction by reacting 0.010 M H2 with 0.010 M I2 at 700 °C. The results are shown in Figure 10.1b. The HI concentration rises to its equilibrium value of 0.015 M, while the H2 and I2 concentrations each drop to their equilibrium value of 0.0025 M. Note that these are the same equilibrium concentrations obtained by the decomposition of HI shown in Figure 10.1a. We conclude that equilibrium concentrations, while dependent upon the initial concentrations, are independent of the direction from which they were obtained.
Figure 10.1b: Concentrations versus Time for the Decomposition of HI at 700 °C
10.1-2. Average Rates
Rates are changes in one quantity with respect to another. For example, speed is the rate of change of position with respect to time. The average speed is determined by dividing the distance traveled (the change in position) by the time required to travel it. Thus, a person traveling 300 kilometers in 4 hours has an average speed of 75 km·h–1 (~50 mph).
change in position
time interval
=
Δx
Δt
=
300 km
4 h
= 75 km·h−1
The above is the average speed over the interval, but it is unlikely that the person traveled at a constant speed of 75 km·h–1 for the entire trip. The instantaneous speed (the speed given by the speedometer at any instant) for such a trip might vary from 0 to over 100 km·h–1.
10.1-3. Average Rates of Formation of HI
Reaction rates are the most important of the kinetic parameters. They are measured as changes in concentration with respect to time. Products are formed during a reaction, so we refer to their rate of formation. The average rate of formation of HI over a time interval Δt is given as the following.
rate of formation of HI =
Δ [HI]
Δt
Consider the following three points in the plot of HI concentration versus time at 700 °C for the reaction H2 + I2→ 2 HI shown in Figure 10.2.
time (s)
[HI] (M)
0
0.000
2000
0.010
5000
0.014
Table 10.1
Figure 10.2
The average rate of formation of HI during the first 2000 s would be determined as
Δ [HI]
Δt
=
0.010 M − 0.000 M
2000 s − 0 s
=
0.010 M
2000 s − 0 s
= 5.0 × 10−6M/s
and the average rate of formation in the interval 2000 to 5000 s would be
Δ [HI]
Δt
=
0.014 M − 0.010 M
5000 s − 2000 s
=
0.004 M
3000 s
= 1 × 10−6M/s
Thus, the rate decreased by nearly 80%. Indeed, the rate of formation continues to decrease as it approaches 0 at equilibrium. Thus, rates of reaction vary with time.
10.1-4. Graphical Determination of a Rate of Formation
It is not the average rate of change that is important in kinetics studies. Rather it is the instantaneous rate of change; i.e., the rate at which the concentration is changing at a given time. The instantaneous rate at a given time is given by the slope of the tangent to the concentration versus time curve at that instant. We will assume that anytime a rate is given, that it is the instantaneous rate unless it is specified to be the average rate.We now determine the rate of formation of HI 2130 s into the reaction. Consider the graph shown in Figure 10.3, which shows the variation of the HI concentration with time in more detail than in Figure 10.1a. The rate of formation of HI at this point equals the slope of the tangent (shown as a green line). The slope is determined by taking two points on the tangent line and determining the change in concentration by the time interval between the two points. We use the two points at the ends of the line to determine the slope and therefore the rate of formation at that time as follows.
rate of formation of HI at 2130 s =
Δ [HI]
Δt
=
+0.008 M
4000 s
= 2 × 10−6M/s
Figure 10.3: Graphical Determination of the Rate of Formation of HI at 2130 s
10.1-5. Graphical Determination of a Rate of Disappearance
Reactant concentrations decrease with time, so they are disappearing. Consequently, it is usually the rate of disappearance of reactants that is given. The rate of disappearance of H2 is given as
rate of disappearance of H2 = −
Δ [H2]
Δt
Note that the rate at which [H2] is changing is negative, so its rate of disappearance is positive.The rates of disappearance of H2 and I2 at 2130 s into the reaction is determined graphically in Figure 10.4. Note that the slope of the tangent is negative, which indicates a disappearance. Thus, the rates of disappearance of H2 and I2 are each 1 × 10–6M·s–1. Note that the rates of disappearance of H2 and I2 are one-half of the rate of formation of HI, consistent with the fact that 2 HI are formed for each H2 and I2 that react.
Figure 10.4: Graphical Determination of the Rates of disappearance of H2 and I2 at 2130 s
10.1-6. Rate of Reaction
The rate of formation of HI at any instant is exactly twice the rate of disappearance of H2 or I2 as dictated by the stoichiometry of the reaction (2 mol HI form for each mole of H2 or I2 that react). The rates of appearance of products and the rates of disappearance of reactants are often different, so what is meant by the term rate of reaction? To answer that question, we construct the reaction table to determine how much reaction takes place during some time interval Δt.
H2
+
I2
→
2 HI
t
c
c
c'
Δt
–Δx
–Δx
+2Δx
t + Δt
c – Δx
c – Δx
c' + 2Δx
In the reaction table, Δx is the change in concentration of any species whose coefficient in the balanced chemical equation is unity. In the reaction above, Δx = –Δ[H2] = –Δ[I2] = Δ[HI]/2. The rate of reaction of the above reaction is defined as
rate of reaction =
Δx
Δt
= −
Δ [H2]
Δt
= −
Δ [I2]
Δt
=
1
2
×
Δ [HI]
Δt
Although it is more common to talk about the rate of formation of a product or the rate of disappearance of a reactant, the rate of reaction is also used and care must be given to the differences caused by the coefficients in the balanced equation. In general, the rate of reaction is related to the rates of formation or disappearance as follows.
( 10.1 )
rate of reaction
=
Δx
Δt
=
rate of formation of product
coefficient of product
=
rate of disappearance of reactant
coefficient of reactant
The Rate of Reaction
10.1-7. Exercise
Exercise 10.1:
At some time during the reaction N2(g) + 3 H2(g) → 2 NH3(g), the rate of formation of ammonia is 0.024 M·s–1. What is the rate of the reaction and what are the rates of disappearance of each of the reactants at this time?
rate of reaction =0.012_0__
rate =
rate of formation of NH3
coefficient of NH3
=
0.024 M/s
2
= 0.012 M
M·s–1
rate of disappearance of N2 =0.012_0__The coefficient of N2 is one, so its rate of disappearance equals the rate of reaction.M·s–1
rate of disappearance of H2 =0.036_0__Solve Equation 10.1 to obtain rate of disappearance. rate of disappearance = (coefficient) × (rate of reaction) = 3 × 0.012 = 0.036 M/sM·s–1
10.1-8. Exercise
Exercise 10.2:
The rate of formation of N2 was found to be 0.32 M/s.
4 NH3(g) + 3 O2(g) → 2 N2(g) + 6 H2O(g)
The rate of reaction at this time was:
0.16_0__
rate of reaction =
rate of formation of N2
coefficient of N2
=
0.32 M/s
2
= 0.16 M/s
M/s
The rate of disappearance of NH3 at that time was:
0.64_0__rate of disappearance of NH3 = (coefficient) × (rate of reaction) = 4 × 0.16 M/s = 0.64 M/sM/s
The rate of disappearance of O2 was:
0.48_0__rate of disappearance of O2 = (coefficient) × (rate of reaction) = 3 × 0.16 M/s = 0.48 M/sM/s
The rate of formation of water was:
0.96_0__rate of formation of H2O = (coefficient) × (rate of reaction) = 6 × 0.16 M/s = 0.96 M/sM/s
10.2. Rate Laws
Introduction
Rates are used to determine the rate law of a reaction, which shows how the rate varies with the concentrations of reactants.
Objectives
•
Determine the rate of a reaction given the rate law, the rate constant, and concentrations of reactants.
•
Determine the rate constant given the rate law and a rate at a given set of concentrations.
10.2-1. Definitions
The manner in which the rate varies at a given temperature is expressed by the rate law for the reaction. Rate laws can be very complicated, but they often take the form of a constant times the concentrations of the reactants each raised to some exponent. The exponent of a reactant's concentration is called the reactant order of that reactant, while the sum of the exponents of all reactants is referred to as the reaction order.
Reaction
Rate Law
Reaction and Reactant Orders
a H2 + I2→ 2 HI
rate = ka[H2][I2]
A second order reaction that is first order in H2 and first order in I2.
b 2 HI → H2 + I2
rate = kb[HI]2
A second order reaction that is second order in HI.
c 2 N2O5→ 4 NO2 + O2
rate = kc[N2O5]
A first order reaction that is first order in N2O5
Table 10.2
It is important to recognize the following.
The exponents in a rate law are not necessarily the stoichiometric coefficients in the balanced equation. The exponents must be determined experimentally.
Thus, Reaction c is first order in N2O5 even though the stoichiometric coefficient of N2O5 is two. The rate constants ka and kb are called second-order rate constants, while kc is a first-order rate constant. The reaction rate has units of concentration per unit time, but the units of the rate constant depend on the reaction order.
First Order
Second Order
kc =
rate
[N2O5]
=
M·s−1
M
= s−1
kb =
rate
[HI]2
=
M·s−1
M 2
= M −1·s−1
Table 10.3
Thus, the units of a first order rate constant are s–1 and the units of a second order rate constant are M–1·s–1.
10.2-2. Exercise
Exercise 10.3:
The rate of decomposition of 0.10 M N2O5 at 298 K is 0.022 M·min–1.
2 N2O5→ 4 NO2 + O2
rate of reaction =0.011_0__Use Equation 10.1,
rate =
0.022 M/min
2
= 0.011 M/min
M·min–1
k298 =0.11_0__
k =
rate
[N2O5]
=
0.011 M/min
0.10 M
= 0.11 min−1
min–1
The reaction continues until the concentration of N2O5 has dropped to 0.042 M. At that point,
rate of reaction =4.6e-3_0__rate = k[N2O5] = (0.11 min–1)(0.042 M) = 4.6e–03 M/minM·min–1
rate of formation of NO2 =0.018_0__Use Equation 10.1,
rate of formation = (coefficient)(rate) = (4)(4.6e−03) = 1.8e−02 M/min
M·min–1
10.3. Determining Rate Laws
Introduction
The exponents of each of the concentrations in a rate law must be determined experimentally. In this section, we apply the method of initial rates and concentration versus time data to obtain rate laws.
Objectives
•
Use the method of initial rates to determine the rate law of a reaction.
10.3-1. Introduction
In the method of initial rates, average rates of reaction are determined at the beginning of the reaction. The time interval of the experiment must be small enough that only a small fraction of the reactants is consumed. This assures that the average rate is close to the instantaneous rate (Δt→ 0). The method has the advantage that the concentrations are initial (makeup) concentrations, which means that they are predetermined. In addition, the initial concentrations of the products are zero, so the reverse reaction can be ignored. This is a real advantage when studying reactions where the forward and reverse reactions compete with one another at appreciable product concentrations.
10.3-2. Method
At least one experiment must be performed for each unknown in the rate law: one for the rate constant and one for each reactant order. For example, the rate law for the generic reaction, A + B → products has the form R = k[A]a[B]b, so a minimum of three rates must be measured using different initial concentrations of A and B in order to determine the values of k, a, and b. The three rates can be expressed as
R1 = k[A]a1[B]b1R2 = k[A]a2[B]b2R3 = k[A]a3[B]b3
If all three experiments are carried out at the same temperature, all three rate constants are the same. Thus, the rate constant can be eliminated by dividing the rates by one another to produce the following two equations.
R1
R2
=
k[A]a1[B]b1
k[A]a2[B]b2
=
[A]1
[A]2
a
[B]1
[B]2
b
and
R2
R3
=
k[A]a2[B]b2
k[A]a3[B]b3
=
[A]2
[A]3
a
[B]2
[B]3
b
We now have two equations in two unknowns that can be solved for a and b by taking the logarithms of both sides. However, the algebra can be simplified by experimental design. If the concentration of B is kept constant in Experiments 1 and 2 ([B]1 = [B]2), and the concentration of A is kept constant in Experiments 2 and 3 ([A]2 = [A]3) then the ratio of rates simplifies to the following.
( 10.2 )
R1
R2
=
[A]a1[B]b1
[A]a2[B]b2
=
[A]1
[A]2
a
and
R2
R3
=
[A]a2[B]b2
[A]a3[B]b3
=
[B]2
[B]3
b
Ratios of Rates
The ratios are all known, so the exponents can be determined with the use of logs as shown in Equation 10.3.
( 10.3 )
a =
log
R1
R2
log
[A]1
[A]2
and b =
log
R2
R3
log
[B]2
[B]3
The Orders of the Reactants
The orders can often be determined by inspection in cases where the ratios are simple fractions. Most of the examples that we will deal with can be done by inspection (no logs required).
10.3-3. Decomposition of HI Exercise
Exercise 10.4:
The kinetics of the decomposition of HI at 700 °C were followed by monitoring the appearance of iodine. Fresh HI was added to the reaction flask, and the times required for the concentration of the iodine to reach 1.00 × 10–4M were determined at different initial concentrations of HI.
Exp
Initial [HI] (M)
Time (s)
1
0.0200
140.3
2
0.0400
35.1
Determine the order of HI and the specific rate constant for the decomposition at 700 °C.
1) Convert the times into rates of formation of I2.
R1 =7.13e-7_0__
rate =
concentration of I2 formed
time required
=
1.00e−4 M I2
140.3 s
= 7.13e−07 M/s
M/s
R2 =2.85e-6_0__
rate =
concentration of I2 formed
time required
=
1.00e−4 M I2
35.1 s
= 2.85e−06 M/s
M/s
2) Determine the ratio of the rates. (Divide the larger by the smaller to avoid fractions.)
R2/R1 =4.00_0_3_
R1
R2
=
2.85e−06 M/s
7.13e−07 M/s
= 4.00
3) Determine the corresponding ratio of concentrations.
and the results of steps 2 and 3 to determine the order of HI. The order is an integer.
order =2_0_1_The ratio of rates = the ratio of concentrations raised to the order 4.00 = (2.00)n, so n = 2
5) Use the rate law you have now determined and one of the concentration–rate pairs to determine the second order rate constant for the reaction.
k =1.78e-3_0__
R = k[HI]2,
so
k =
Rate
[HI]2
Substituting data from Experiment 1:
k =
7.13e−07 M/s
(0.0200 M)2
= 1.78e−03 M−1·s−1
M–1·s–1
10.3-4. Formation of HI Exercise
Exercise 10.5:
The kinetics of the formation of HI at 700 °C were followed by measuring the time required for the concentration of the iodine to drop by 1.00 × 10–4M.
Exp
[H2] (M)
[I2] (M)
Time (s)
1
0.0100
0.0100
16.0
2
0.0200
0.0100
8.00
3
0.0200
0.0200
4.00
Determine the order of H2 and I2 and the specific rate constant for the formation of HI at 700 °C.
1) The rate of reaction equals the rate at which I2 disappears, so we must first convert the times into rates of disappearance of I2.
R1 =6.25e-6_0__
rate =
concentration of I2 disappears
time required
=
1.00e−4 M I2
16.0 s
= 6.25e−06 M/s
M/s
R2 =1.25e-5_0__
rate =
concentration of I2 disappears
time required
=
1.00e−4 M I2
8.00 s
= 1.25e−05 M/s
M/s
R3 =2.50e-5_0_3_
rate =
concentration of I2 disappears
time required
=
1.00e−4 M I2
4.00 s
= 2.50e−05 M/s
M/s
2) Pick the best ratio of rates to use to determine the order of H2.
R2/R1
R3/R2To determine the order of H2, take the ratio of rates in experiments where [H2] changes, but [I2] does not.
R3/R1To determine the order of H2, take the ratio of rates in experiments where [H2] changes, but [I2] does not.
3) Determine the ratio of the rates to use to determine the order of H2.
ratio =2.00_0_3_
R2
R1
=
1.25e−05 M/s
6.25e−06 M/s
= 2.00
4) Determine the corresponding ratio of concentrations.
and the results of Steps 7 and 8 to determine the order of I2.
order of I2 =1_0_1_The ratio of rates = the ratio of concentrations raised to the order 2.00 = (2.00)n, so n = 1
10) Use the rate law you have now determined and the data from one experiment to determine the rate constant for the reaction.
k =0.0625_0__
R = k[H2][I2],
so
k =
Rate
[H2][I2]
Substituting data from Experiment 1:
k =
6.25e−6 M/s
(0.0100 M)(0.0100 M)
= 6.25e−02 M−1·s−1
M/s
10.3-5. Exercise
Exercise 10.6:
The initial rates data at some temperature for the reaction
NO(g) + H2(g) → N2O(g) + H2O(g)
are given in the following table.
Exp
[NO] (M)
[H2] (M)
Rate (M/min)
1
0.10
0.80
0.26
2
0.30
0.80
2.34
3
0.30
0.40
1.17
The order of NO is
2_0_1_
R2
R1
=
2.34
0.26
= 9.0
[NO]2
[NO]1
=
0.30
0.10
= 3.0
Thus, 3.0n = 9.0, so n = 2 and the reaction is second order in NO.
The order of H2 is
1_0_1_
R2
R3
=
2.34
1.17
= 2.00
[H2]2
[H2]3
=
0.80
0.40
= 2.0
Thus, 2.0n = 2.0, so n = 1 and the reaction is first order in H2.
The specific rate constant at this temperature is
k =32.5_0.5__
R = k[NO]2[H2],
so
k =
R
[NO]2[H2]
Substitute data from Experiment 2 to obtain
k =
2.34 M/min
(0.30 M)2(0.80 M)
= 32.5 M−2·s−1
M–2·s–1
First-order Kinetics
10.3-6. Integrated Rate Law
Consider the generic first order reaction A → products, which has the following rate law.
−
Δ[A]
Δt
= k[A]
The above can be rearranged as follows to get the [A] terms on the same side of the equation
−
Δ[A]
[A]
= kΔt
which can be solved by the following integration
[A]
d[A]
[A]
[A]0
= −k
t
dt
0
to produce the following two expressions that relate concentration and time.
( 10.4 )
ln [A] = ln [A]0 − kt
ln
[A]
[A]0
= −kt
The Integrated Rate Equation for a First Order Reaction
where [A]0 is the concentration of A at t = 0, i.e., the initial concentration. Equation 10.4 shows that a plot of ln [A] versus time for a first order reaction is a
straight line with a slope of –k and an intercept of ln [A]o.This behavior can also be expressed as an exponential.
( 10.5 )
[A] = [A]0e−kt
Exponential Form of the Integrated Rate Equation
Equation 10.5 shows that the concentration of a reactant drops exponentially with time. This behavior is called exponential decay. We conclude the following.
A reactant in a first order reaction undergoes exponential decay, and a plot of ln [A] vs. t for its decay is linear with a slope of –k.
10.3-7. Determining a Concentration Exercise
Exercise 10.7:
The decomposition of dimethyl ether,
H3C−O−CH3(g) → CH4(g) + H2(g) + CO(g)
is first order with a rate constant of 4.00 × 10–4 s–1 at 500 °C. If the initial concentration of dimethyl ether is 0.050 M, what is its concentration after 1.00 hour?
[HC3–O–CH3[ =0.012_0_2_Use Equation 10.5 with the given initial concentration, rate constant, and elapsed time. Exponentials must be unitless, so that the elapsed time must be in seconds because the rate constant is in s–1, i.e., kt is unitless. [A] = [A]0e–kt = (0.050)e–(4.00 × 10–4)(3600) = 0.050e–1.44 = 0.012 MM
10.3-8. First Order Behavior Exercise
Exercise 10.8:
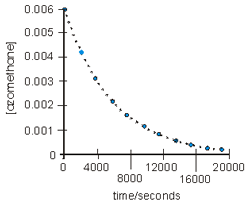
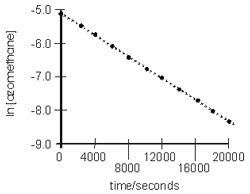
The concentrations of azomethane at various times during its decomposition at 300 °C are shown in the table below. Verify that the decomposition is first order and determine the first-order rate constant at 300 °C.
The decomposition reaction is
H3C−N=N−CH3(g) → C2H6(g) + N2(g).
t (s)
[A]
ln [A]
[A] vs t
ln [A] vs t
0
0.00600
–5.116
2000
0.00436
–5.436
4000
0.00316
–5.756
6000
0.00230
–6.076
8000
0.00167
–6.396
10000
0.00121
–6.716
12000
0.00088
–7.036
14000
0.00064
–7.356
16000
0.00046
–7.676
18000
0.00034
–7.996
20000
0.00024
–8.316
The reaction is first order because a plot of ln [A] versus t is linear.
What is the first order rate constant?
k =1.6e-4_0__The slope of ln [A] vs. t equals –k, so determine the slope of the line. All of the points lie on the line, so the slope can be determined by using any two points as follows.
slope =
ln [A]2 − ln [A]1
t2 − t1
Substitution of the t1 = 4000 s and t2 = 16000 s points, we obtain the following.
slope =
−7.676 − (−5.756)
16000 s − 4000 s
=
−1.92
12000 s
= −1.60e−4 s−1
The rate constant is –slope, so k = 1.60e–4 s–1.s–1
10.3-9. Half-life Definition
The half-life, t1/2, of a reaction is the time required for half of the existing reactant to disappear. Consider the graph shown in Figure 10.5. The initial concentration of A (azomethane) is 0.006 M, so the half-life is the time required for the concentration to drop to 0.003 M. As shown in the figure, this occurs after 4330 s, the half-life of the reaction. After another half-life (another 4330 s) the concentration has dropped to 0.0015 M, which is half of 0.003 M. After a third half-life the concentration is again halved. Thus the half-life of a first order reaction is constant.The concentration of a reactant after n half-lives is given by the following.
[A] =
[A]0
2n
Thus, after five half-lives (5 × 4330 = 21,650 s) the concentration of azomethane is (1/2)5(0.006 M) = 1.9 × 10–4M.
Figure 10.5: The Half-life of Azomethane
The half-life of the decomposition of azomethane at 300 °C is 4330 seconds. Thus, the concentration is halved every 4330 seconds.
10.3-10. Half-life Equation
At the half-life, t = t1/2 and [A] = [A]0/2. Substitution of these half-life quantities into Equation 10.4
Note that the identity ln(1/x) = –ln(x) was used. Solving for the half-life,
( 10.6 )
t1/2 =
ln 2
k
=
0.693
k
The Half-life of a First Order Reaction
The half-life of the azomethane decomposition can be determined from its first order rate constant determined in Exercise 10.8
(k = 1.60 × 10−4 s−1)
with Equation 10.6 as follows
t1/2 =
0.693
1.60 × 10−4
= 4330 s
which is the same value observed in the previous section. As shown in the following exercise, Equation 10.6 can also be used to determine the value of the first-order rate constant from a half-life.
10.3-11. Half-life Exercise
Exercise 10.9:
If a first-order reaction has a 23.5 minute half-life, how long would it take for it to reach 90.0% completion?
k =0.0295_0__Use Equation 10.6 to determine the rate constant from the half-life.
k =
ln 2
t
=
0.693
23.5 min
= 0.0295 min−1
min–1
[A]/[A]0 =0.100_0_3_100% of the compound is present initially ([A]0), and 90.0% completion means that 90.0% A reacts, so only 10.0% ([A]) remains.
[A]
[A]0
=
10.0%
100%
= 0.100
t =78.1_0__Use the rate constant and concentration ration determined above in Equation 10.4 to determine the required time.
t =
ln([A]/[A]0)
k
=
ln(0.100)
0.0295
= 78.1 min
min
Second-order Kinetics
10.3-12. Integrated Rate Law
Consider the generic second order reaction 2 A → products, which has the following rate law.
−
Δ[A]
Δt
= kΔ[A]2
The above can be rearranged as follows to get the [A] terms on the same side of the equation,
−
Δ[A]
[A]2
= kΔt
which can be solved by the following integration
−
[A]
d[A]
[A]2
[A]0
= k
t
dt
0
to produce the following expression that relates concentration and time.
( 10.7 )
1
[A]
=
1
[A]0
+ kt
The Integrated Rate Equation for a Second Order Reaction
Thus a plot of 1/[A] vs. t for a second order reaction is a straight line with an intercept of 1/[A]0 and a slope equal to the rate constant.
10.3-13. Reaction Order Exercise
Exercise 10.10:
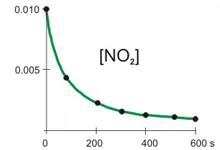
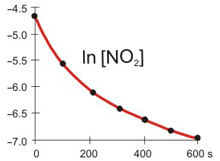
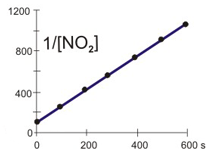
The concentration of NO2 versus time during its decomposition at 350 °C is shown in the table below. The natural logarithms and reciprocals of the concentrations are also included in the table. Determine if the decomposition is first or second order and the rate constant for the decomposition at 350 °C.
The decomposition reaction is
2 NO2(g) → 2 NO(g) + O2(g).
t (s)
[NO2]
ln [NO2]
1/[NO2]
0.0
0.0100
–4.605
100
100.0
0.0038
–5.565
261
200.0
0.0024
–6.045
422
300.0
0.0017
–6.368
583
400.0
0.0013
–6.612
744
500.0
0.0011
–6.808
905
600.0
0.0009
–6.972
1066
Plots
Refer to the plots above to view the kinetics of the decomposition of NO2 graphically. Then determine the NO2 order in the decomposition.
Is this a first or second order reaction?
first order reactionThe plot of ln [A] vs. t is not linear, so the reaction is not first order in A.
second order reaction
The rate constant for the rate of disappearance of NO2 is:
k =1.61_0__The slope of 1/[A] vs. t equals k. All points lie on the line, so choose any two to obtain the slope.
k = slope =
1/[A]2 − 1/[A]1
t2 − t1
=
744 M−1 − 261 M−1
400.0 s − 100.0 s
=
483 M−1
300.0 s
= 1.61 M−1·s−1
M–1·s–1
The rate constant for the rate of reaction is:
k =0.805_0__
rate constant for reaction =
rate constant for disappearance
coefficient
=
1.61
2
= 0.805 M−1·s−1
M–1·s–1
Another Look at the Relationship Between Equilibrium and Kinetics
10.3-14. Decomposition of HI Review
At equilibrium, the rates of the forward and reverse reactions are equal.
Recall that HI decomposes into H2 and I2, but it can also be formed from H2 and I2. If we represent these competing processes as 2 HI ⇌ H2 + I2 then the decomposition of HI is the forward reaction with a rate constant kf, and the formation of HI is the reverse reaction with a rate constant kr. We studied the competing reactions using the method of initial rates. The resulting rate laws were the following.
2 HI → H2 + I2
−
Δ[HI]
Δt
= 2kf[HI]2
H2 + I2→ 2 HI
Δ[HI]
Δt
= 2kr[H2][I2]
The specific rate constants at 700 °C were found to be kf = 1.8 × 10–3M–1·s–1 and kr = 6.3 × 10–2M–1·s–1. The '2' in the rate laws is due to the coefficient of HI in the balanced equation.
10.3-15. Deviation from Second Order Kinetics
Figure 10.6: A plot of 1/[HI] versus time for the decomposition of HI at 700 °C
The reaction is second order in HI, but the plot is not linear at later times because of competition from the reverse reaction.
The decomposition of HI is second order in HI, so a plot of 1/[HI] versus time for the decomposition should be linear. As shown in Figure 10.6, the plot is linear at the beginning of the experiment, but it deviates from linearity after about 2000 s. The curvature of the line indicates that [HI] is greater than predicted by simple second order kinetics. The higher than expected concentration of HI results because it is being formed by the formation reaction. As the reaction proceeds, the decomposition slows because the concentration of HI drops, but the rate of formation increases
because the concentrations of H2 and I2 are increasing. The net rate of disappearance of HI is equal to the rate at which it disappears in the decomposition minus the rate at which it forms in the formation. At equilibrium, the concentration of HI no longer changes because it is formed at the same rate that it disappears. Thus, at equilibrium,
rate of appearance of HI
=
rate of disappearance of HI
kf[HI]2
=
kr[H2][I2]
Gathering concentrations on one side and rate constants on the other, and substituting the known rate constants, we obtain the following:
kf
kr
=
1.8e−3 s−1
6.3e−2 s−1
= 0.029 =
[H2][I2]
[HI]2
= Kc
which is the equilibrium constant for the reaction 2 HI(g) ⇌ H2(g) + I2(g). We conclude that the equilibrium constant is equal to the ratio of the forward and reverse rate constants. The equilibrium concentrations in the decomposition of 0.020 M HI at 700 °C are [HI] = 0.015 M, and [H2] = [I2] = 0.0025 M. Using the known equilibrium concentrations and rate constants, we can determine the rates of formation and disappearance of HI as follows.
rate of disappearance = 2kr[I2][H2] = 2(6.3 × 10–2M–1·s–1)(0.0025 M)(0.0025 M) = 8.0 × 10–7M/s
The reaction reaches equilibrium because HI is formed at the same rate that it is consumed. Thus, while the net concentrations are no longer changing at equilibrium, the reaction has not stopped.
10.4. Reaction Mechanisms and Rate Laws
Introduction
The rate law of a reaction gives us some clues about the steps involved in the reaction. In this section, we examine some simple examples.
Objectives
•
Identify the intermediates in a reaction mechanism and use an experimental rate law to identify the rate-determining step.
•
Determine the molecularity of an elementary step.
•
Determine if a mechanism is consistent with the rate law.
10.4-1. Elementary Reactions
Most reactions do not occur in a single step, but by a series of elementary steps called the reaction mechanism. Consider the decomposition of azomethane,
H3C−N=N−CH3→ C2H6 + N2
which proceeds by a two-step mechanism.
1
H3C–N=N–CH3→ 2 CH3 + N2
2
2 CH3→ C2H6
In the first step, the two N–C bonds break and the N≡N bond forms, and in the second, the highly reactive CH3 groups quickly combine to form ethane. The individual steps of the mechanism are called elementary reactions because they each represent a single molecular event that results in a reaction. The two elementary reactions above combine to give the reaction mechanism for the decomposition of azomethane. The sum of the elementary processes must yield the net reaction. The two CH3 (methyl) groups that are formed in the first step are consumed in the second. Since they appear in the mechanism, but not in the net reaction, the methyl groups are called intermediates. Intermediates typically are short-lived, but they can frequently be observed during reaction and sometimes even isolated. Indeed, evidence for a proposed intermediate is excellent support for a mechanism.
10.4-2. Molecularity
The number of molecules reacting in an elementary reaction is called the molecularity of the elementary reaction. There are only three molecularities.
Number of Particles
Molecularity
1
unimolecular
2
bimolecular
3
termolecular
Termolecular processes are very rare because the simultaneous collision of three reactant molecules is very rare. Processes with molecularities greater than three do not occur at all. Thus, the first step in the decomposition of azomethane is unimolecular while the second step is bimolecular.
10.4-3. The Rate Law of an Elementary Process
In order for two or three molecules to react, they must collide with one another. As a result, the rate of an elementary reaction is proportional to the collision frequency of the reactants. The collision frequency, which is the number of collisions between the reacting particles in a specified volume per unit time, can be shown to be directly proportional to the product of the molar concentrations of the colliding particles. Thus, we conclude the following.
The rate law of an elementary reaction is equal to a proportionality constant (the rate constant) times the product of the molar concentrations of the reactants.
This means we can write the rate law expression for elementary processes from their chemical equation. For example,
A + B → C rate = k[A][B]
2A → D rate = k[A][A] = k[A]2
Most reactions do not occur in a single step, so their rate laws cannot be determined from the overall balanced equation. They can be determined only by experiment.
10.4-4. Rate-Determining Step
The rate law of a reaction is a combination of the rate laws of the elementary reactions of which it is composed. Thus, mechanisms are proposed based on the observed rate law. A mechanism is acceptable only if the rate law derived from it agrees with the experimentally determined rate law. It is often the case that more than one mechanism yields the experimental rate law, so mechanisms are usually proposed, not proven. However, finding evidence for a proposed intermediate strongly supports a proposed mechanism and can sometimes prove that it is correct. Combining the rate laws of the elementary processes into a reaction rate law can be a formidable task for complicated reactions, so we restrict our discussion to a special case: reactions in which one step of the multi-step mechanism is much slower than any of the others. In this case,
•
the rate of reaction is determined by the rate of the slow step, which is called the rate-determining step (RDS), and the rate law of the reaction is the rate law of the RDS.
10.4-5. Rate-Determining Step Example
Example: As a simple example of a process with a rate-determining step, consider one in which three workers are constructing chairs in an assembly line. The process involves combining four legs (L), two arms (A), one back (B), and a seat (S) to make a chair (L4A2SB): 4 L + 2 A + B + S → L4A2SB. The chairs are assembled in a three-step process:
1
S + B → SB
2
SB + 4 L → L4SB
3
L4SB + 2 A → L4A2SB
The SB units, which are intermediates, are made at a rate of 60 an hour and placed in a box for the next worker. The second worker attaches the four legs, which takes longer to do, and he can only produce 20 of the L4SB units per hour.The final worker can make 40 chairs an hour if all of the components are available, but the L4SB units arrive at a rate of only 20 an hour. Consequently, the final worker makes a chair and then waits for the second worker to put another unit in the box. The third step cannot proceed any faster than the second step for lack of supplies. The first worker starts making units at a rate of 60 an hour and puts them in the box, but they are removed from the box at a rate of only 20 an hour. Consequently, the box gets full and the first worker must wait for the second worker to remove a unit before a new SB unit can be made. The first step has reached equilibrium, and the rate at which SB units are produced is limited by the rate at which the second worker removes units from the box. The rates of the two rapid steps are limited by the slow (rate-determining) step, so only 20 chairs an hour can be produced in this assembly line.
10.4-6. Intermediates and the Rate Law
The rate law of a reaction is based on the rate law of the rate-determining step, but the rate laws of reactions considered in this chapter involve the concentrations of only reactants—no products and no intermediates. Consequently, the concentrations of any intermediates that appear in the rate law of the RDS must be eliminated to obtain the experimental rate law. This is done by assuming that the rapid steps that precede the rate-determining step all reach equilibrium. Thus, the concentrations of any intermediate can be obtained in terms of the concentrations of the reactants and the equilibrium constants of the preceding reactions. This results in the following two properties of the rate law.
•
The rate constant of a reaction is composed of the rate constant of the RDS and equilibrium constants of previous steps.
•
The rate law of a reaction contains only those reacting molecules that appear in the RDS and steps prior to the RDS.
10.4-7. Determining a Rate Law from a Mechanism
Let us determine the rate law for the reaction with the following mechanism.
1
NO + O2→ NO3
2
NO + NO3→ 2 NO2
The rate law can contain only the concentrations of reactants in the net equation, so we begin by writing the net equation. NO3 is an intermediate that is formed in Step 1 and consumed in Step 2, so the net equation is:
2 NO + O2→ 2 NO2
Only NO and O2 appear as reactants in the net equation, so only they can appear in the rate law. The rate law of the reaction is the rate law of the RDS. We will do this problem twice, assuming each step to be rate-determining. Remember that the rate law of an elementary process equals the rate constant times the concentrations of the reactants each raised to an exponent equal to their coefficients in the balanced equation.If Step 1 is RDS: NO + O2→ NO3 is the RDS, so the rate law of the RDS is rate = k[NO][O2]. The rate law of the RDS contains the concentrations of reactants in the net equation only, so this result would be acceptable for the rate law of the reaction.If Step 2 is RDS: NO + NO3→ 2 NO2 is the RDS, so the rate law of the RDS is
rate = k' [NO][NO3].
NO3 is an intermediate, so it cannot appear in the rate law. To eliminate NO3, we assume that the first (rapid) step reaches equilibrium to get an expression for [NO3] in terms of reactant concentrations. The equilibrium constant expression for the first reaction is
K =
[NO3]
[NO][O2]
solving for the NO3 concentration, we obtain
[NO3] = K[NO][O2]
Substitution of this expression into the rate law for the RDS, yields the following.
rate = k' [NO]{K[NO][O2]} = k'K[NO]2[O2]
Substituting k for
k'K
, we obtain the rate law expression.
rate = k[NO]2[O2]
The result contains the concentrations of only reactants in the net equation, so it would be acceptable for the rate law of the reaction. Note that the rate law of the reaction is equal to the product of the rate constant of the RDS and the equilibrium constant of the previous step. We could arrive at the same rate laws by using the rule that the concentrations of only those molecules involved in the RDS and prior steps appear in the rate law. Thus, only one NO appears in the rate law if the first step is rate-determining because the second NO does not appear until the second step.
10.4-8. Rate Law from Mechanism Exercise
Exercise 10.11:
The reaction, NO2(g) + CO(g) → NO(g) + CO2(g) is believed to occur by the following two-step mechanism.
1
2 NO2(g) → NO3(g) + NO(g)
2
NO3(g) + CO(g) → NO2(g) + CO2(g)
What is the molecularity of Step 1?
unimolecular Two molecules of NO2 are involved.
bimolecular
termolecular Two molecules of NO2 are involved.
What is the molecularity of Step 2?
unimolecular Two molecules (1 NO3 + 1 CO) are involved.
bimolecular
termolecular Two molecules (1 NO3 + 1 CO) are involved.
Which of the following is an intermediate?
NO2Intermediates do not appear in the net equation.
CO Intermediates do not appear in the net equation.
NO3
CO2Intermediates do not appear in the net equation.
Assume that Step 1 is the RDS to determine the reactant orders.
order of NO2: 2_0__Two molecules of NO2 are involved in the RDS and there are no previous steps, so the reaction is second order in NO2.
order of CO: 0_0__No molecules of CO are involved in the RDS and there are no previous steps, so the reaction is zero order in CO. This means that the rate of reaction is independent of the CO concentration.
Assume that Step 2 is the RDS to determine the reactant orders.
order of NO2: 2_0__No NO2 molecules are involved in the RDS, but two molecules are involved in the previous step.
order of CO: 1_0__One CO molecule is involved in the RDS, but none are involved in the previous step.
10.4-9. A Three-step Mechanism Exercise
Exercise 10.12:
The reaction of nitric oxide with hydrogen, 2 NO(g) + 2 H2(g) → N2(g) + 2 H2O(g) is believed to proceed by the following three-step mechanism, which is consistent with the experimental rate law.
1
2 NO ⇌ N2O2 rapid equilibrium with equilibrium constant K1
Use the above mechanism to determine the order of each reactant.
order of NO: 2_0__0NO is not involved in the RDS, but two molecules of NO are involved prior to the RDS.
order of H2: 1_0__One molecule of H2 is involved in the RDS, but the other is not involved until after the RDS.
10.5. The Effect of Temperature on Reaction Rates
Introduction
The rate of reaction depends upon both the reactant concentrations and the temperature. We have discussed the concentration dependence in the rate law, and we now treat the temperature dependence.
Prerequisites
•
CAMS 9.9 Activation Energy
Objectives
•
Use the Arrhenius equation to solve for a variable.
•
Sketch a potential energy profile for a reaction and interpret it in either the forward or reverse direction.
•
Define reaction coordinate, transition state, and activation energy.
10.5-1. Activation Energy
The rate of reaction depends upon both the reactant concentrations and the temperature. We have discussed the concentration dependence and now treat the temperature dependence. Consider the reaction between methyl iodide and hydroxide ion.
ICH3 + OH1−→ I1− + H3COH
The reaction is an elementary process, so it is first order in each reactant. In order for the reaction to occur, a hydroxide ion must collide with a CH3I molecule, and it must collide between the three hydrogen atoms. As the distance between the hydroxide ion and the carbon atom decreases, the carbon-iodine bond stretches, and the three hydrogen atoms are pushed back, which forces the H–C–H angles to increase from 109°. As shown in Figure 10.7, these changes result in an energy increase, which reaches a maximum when the bond angle is 120° and the carbon is five-coordinate.
Figure 10.7: Energy diagram for CH3I + OH1–→ CH3OH + I1–
The five-coordinate species (at the top of Figure 10.7) is called the transition state of the reaction because the reaction must proceed through this state to make the transition from reactants to products. The transition state can lead to or be formed from either side of the reaction (iodide ion can collide with CH3OH to lead to the same transition state). The transition state cannot be isolated or directly observed. It is not an intermediate but rather a highly energetic species through which the reaction proceeds. The energy required to reach the transition state is called the activation energy. Figure 10.7 shows that the variation of energy along a complicated combination of C–I and C–O bond lengths and H–C–H bond angles is called the reaction coordinate. The activation energy of the forward reaction, Ea(f), is much less than that of the reverse reaction, Ea(r), because the energy of the reactants is greater than the energy of the products; i.e., the reaction is exothermic.
10.5-2. Activation Energy Factors
There are two factors that dictate whether a collision achieves the transition state: the steric factor and the energy factor. The steric factor indicates the fraction of collisions in which the reactants have the correct orientation to react. As shown in Figure 10.8, there are a number of ways in which CH3I and OH1– can collide, but only a fraction have the correct orientation to reach the transition state.
Figure 10.8: Steric Factor
Collisions like (a) and (b) cannot lead to the transition state because the reactants are not aligned properly. Collision (c) can lead to the transition state but only if the colliding particles have sufficient energy to overcome the activation energy.
The energy factor arises because even if the reactants have the correct orientation to react, they cannot do so unless there is sufficient energy in the collision to overcome the activation energy and achieve the transition state. The average energy available in a collision depends on the thermal energy (~RT), and the fraction of collisions with sufficient energy to overcome the activation energy is given by
e−Ea/RT.
The rate constant represents the fraction of collisions that lead to the transition state, and it is the product of the steric and energy factors as shown in Equation 10.8.
( 10.8 )
k = Aexp
−
Ea
RT
= Ae−Ea/RT
Arrhenius Equation
A, which is referred to as the pre-exponential, includes the steric factor. Equation 10.8, known as the Arrhenius equation, shows the temperature dependence of the rate constant. Thus, as the temperature increases, the thermal energy increases, and the fraction of collisions with sufficient energy to achieve the transition state increases. Increasing the temperature, however, increases the rate of both the forward and reverse reactions.
10.5-3. Arrhenius Plot
If a plot of ln k versus 1/T is linear for a reaction, then the reaction displays Arrhenius behavior.
If we take the natural logarithm of both sides of Equation 10.8
, we can rewrite the Arrhenius equation as shown in Equation 10.9.
( 10.9 )
ln k = ln A −
Ea
R
·
1
T
Logarithmic Form of the Arrhenius Equation
A plot of ln k vs. 1/T is called an Arrhenius plot. If a reaction follows Arrhenius behavior, then the plot is linear with slope = –Ea/R and intercept = ln A.An Arrhenius plot is an excellent way to obtain the activation energy of a reaction.
10.5-4. Arrhenius Exercise
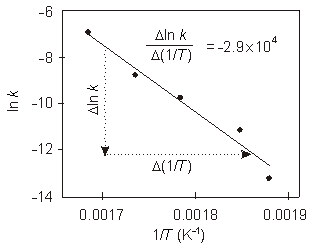
Exercise 10.11:
The following rate constants have been measured for the decomposition of azomethane.
T (K)
k (s–1)
1/T (K–1)
ln k
ln k vs. 1/T
1
532
1.8e–6
0.00188
–13.23
2
541
1.5e–5
0.00185
–11.11
3
560
6.0e–5
0.00179
–9.72
4
576
1.6e–4
0.00174
–8.74
5
593
9.5e–4
0.00169
–6.96
What is the activation energy for the reaction?
E =240_0_2_Ideally, a least squares fit of the data should be done to obtain the best slope. The least square squares result is shown in the figure to be –2.9e+04 K. However, if you do not have that capability on your calculator, use two data points that best describe the slope of the best line to obtain the slope. In this problem, we will use the first and third points.
slope =
Δ ln k
Δ (1/T)
=
−6.96 − (−9.72)
0.00169 − 0.00179
= −2.8e04 K
The fact that the result obtained from Points 3 and 5 is different from the least squares result indicates that these two points do not lie on the line. We use the least squares result to obtain the answer: Ea = –R(slope) = –(8.314 J·K–1·mol–1)(–2.9e04 K) = 2.4e+05 J/mol = 240 kJ/molkJ/mol
10.5-5. Using Only Two Temperatures
Determining the activation energy graphically is the best procedure, but it is often the case that only two data points are known. In these cases, we need consider only two temperatures: T1 and T2 and the two rate constants, k1 and k2, at those temperatures. In addition, only two temperatures are needed to determine a rate constant at one temperature given the activation energy and rate constant at another temperature.To get the expression for only two temperatures, apply Equation 10.9
to each point and then take the difference to eliminate the ln A term and obtain the following.
( 10.10 )
ln k2 − ln k1 = ln
k2
k1
=
Ea
R
1
T1
−
1
T2
Determining One Rate Constant from Another
Equation 10.10 is an excellent way to determine a rate constant at one temperature given the rate constant at another temperature and the activation energy. However, we can also solve the expression for the activation energy, which allows us to determine the activation energy given the rate constants at two temperatures.
( 10.11 )
Ea =
R ln
k2
k1
1
T1
−
1
T2
Activation Energy from the Rate Constants at Two Temperatures
However, care must always be taken when assuming a linear relationship with only two points. The activation energies derived from the above expression can deviate substantially from that determined from the best fit when the two points do not lie on the best line.
10.5-6. Activation Energy Exercise
Exercise 10.14:
The rate constants for the decomposition of azomethane at 532 and 593K are 1.8e–06 s–1 and 9.5e–04 s–1, respectively. Estimate the activation energy of the decomposition from this data.
Ea =270_0_2_
Ea =
(8.314 J/mol·K)ln
9.5e−04 s−1
1.8e−06 s−1
1
532 K
−
1
593 K
= 2.7e+05 J/mol = 270 kJ/mol
kJ/mol
What would the rate constant be at 550 K?
k =1.3e-5_0__Substitute T1 = 532 K, k1 = 1.8e–06 s–1, Ea = 2.7e05 J/mol, and T2 = 550 K into Equation 10.10 to obtain
ln k2 − ln(1.8e−06) +
2.7e05
8.314
×
1
532
−
1
550
= −11.23
Therefore, k2 = e–11.23 = 1.3 × 10–5 s–1.s–1
10.5-7. Activation Energy from Ratio Exercise
Exercise 10.15:
What is the activation energy of a reaction, if its rate at 75 °C is four times what it is at 25 °C?
E =24_0__
Ea =
8.314 J/mol·K × ln
4k298
k298
1
298 K
−
1
348 K
= 2.4 × 104 J/mol = 24 kJ/mol
kJ/mol
10.6. Catalysis
Introduction
Catalysts increase the rate of reaction but are unchanged by it. They function by altering the path (mechanism) of the reaction so as to reduce the activation energy.
Prerequisites
•
CAMS 14.5 Metals as Catalysts
Objectives
•
Explain how the addition of a catalyst affects the reaction rate.
10.6-1. Review
Heterogeneous catalysts are in a different phase (usually solid) than the reactants. Look at the material listed as a prerequisite for this section for a discussion of the catalytic converter and the surface catalyzed hydrogenation of ethene to ethane. Homogeneous catalysts are in the same phase as the reaction. (See the discussion of the titanocene catalyzed polymerization of ethene discussed in the prerequisite.) Catalysts increase the rates of both the forward and reverse reactions, but they do not affect the equilibrium constant. Thus, they increase the rate at which equilibrium is established but not the amount of product that is formed. Catalysts not only increase the rate of reaction, they also lower the temperature required for reaction. Catalysts can also be very specific, increasing the rate of desired reactions without affecting the rates of undesired reactions. Enzymes are very specialized catalysts used by biological organisms.
10.6-2. Ozone's Role in the Stratosphere
High-energy ultraviolet (UV) light (E = hν) from the sun can damage living tissue because its energy is sufficient to break bonds in DNA, thus damaging genes. Fortunately, UV radiation is absorbed by ozone (O3) in the stratosphere:
O3 + hν→ O2 + O
O can go on to react with another O2 to regenerate the ozone, which helps maintain the ozone level in the stratosphere. However, the oxygen atom can also react with an ozone molecule as shown below.
( Reaction 1 )
O3 + O → 2 O2
This reaction would deplete the ozone level, but it has a fairly high activation energy (17 kJ/mol) and few collisions in the stratosphere result in reaction.CFC's (chlorofluorocarbons), which are used as air conditioner refrigerants and in the production of plastics, are very stable and relatively unreactive. However, they absorb high-energy photons in the stratosphere to produce chlorine atoms. Consider the photochemical (caused by light) decomposition of CF2Cl2 (Freon-12):
CF2Cl2 + hν→ CF2Cl + Cl
The Cl atoms produced in reactions of this type can go on to catalyze the reaction between O and O3. The behavior is discussed in the following section.
10.6-3. Ozone Depletion
The Cl atoms formed in the photochemical decomposition of CFC's can go on to catalyze the reaction between O and O3 by the following two-step mechanism.
1
O3 + Cl → OCl + O2
2
OCl + O → Cl + O2
The activation energy for Step 1 is only 2.1 kJ/mol, so it proceeds quite rapidly. The energy diagram for the reaction is given in the figure below. Summing Steps 1 and 2 yields Reaction 1 given in the previous section, with Cl forming OCl as an intermediate. Catalytic Cl lowers the activation energy for the reaction from 17 kJ/mol down to 2 kJ/mol and dramatically speeds the reaction, but it does not affect the initial or final states, so it does not affect the thermodynamics and equilibrium constant of the reaction.
Figure 10.9: Catalytic Cl Lowers the Activation Energy from 17 to 2 kJ/mol
Energy diagram for the reaction O3(g) + O(g) → 2 O2(g) (a) in the absence and (b) in the presence of catalytic chlorine atoms (blue curve). The activation energy
E'a
is 2.1 kJ/mol. Note that OCl lies in a shallow well between the reactants and the products, which is typical of intermediates, while the transition states O3–O, O3–Cl, and O–Cl all lie at peak maxima.
10.7. Exercises and Solutions
Select the links to view either the end-of-chapter exercises or the solutions to the odd exercises.








![line graph [azomethane] versus time in seconds, denoting half-lives](images/figure10-6.png "Figure 10.5: line graph [azomethane] versus time in seconds, denoting half-lives")